Recommended
1. Motivation and Context
While oral antivirals are not a substitute for vaccination, they are key to preventing severe illness, saving lives, and preserving health systems. Vaccination rates continue to be low in low-income countries; only 14 percent of the population in low-income countries has received one dose of the vaccine, compared to 80 percent in high- and upper-middle-income countries.[1] mRNA vaccines, which seem to offer the best protection against severe disease in most groups,[2] are even more concentrated in the wealthiest countries than overall vaccines. As vaccination rates lag and uncertainties about future variants persist, easy-to-administer oral antiviral medications can play an increasingly important role in tackling COVID-19. While treatments like monoclonal antibodies and remdesivir have proven to be efficacious, they require administration via infusion or injection, adding complexity and costs that oral antivirals circumvent.[3]
In December 2021, the FDA granted emergency use authorizations for two oral antiviral treatments against COVID-19: a nirmatrelvir/ritonavir combination (Paxlovid) developed by Pfizer and molnupiravir developed by Merck. Pfizer and Merck signed early voluntary licensing agreements with the Medicines Patent Pool (MPP) covering around 100 low- and middle-income countries (LMICs) (details below for each), but several gaps remain to be filled for this mechanism to translate into adequate supply for LMIC use. Selection of licensees by MPP has been announced for both nirmatrelvir/r[4] and molnupiravir.[5] Translating licenses into commercial-grade production will require incentivizing new investments into manufacturing capacity by licensed manufacturers, securing robust sources of active pharmaceutical ingredients (APIs) and starting materials, ensuring compliance with regulatory standards, and developing distribution agreements, among other milestones. Some of these steps also apply to direct voluntary licensing agreements with generic manufacturers—a strategy pursued by Merck with five generic manufacturers in India for molnupiravir.[6]
As the US continues its effort to fight the pandemic globally through a coordinated, whole-of-government response, COVID-19 treatments are key to saving lives and lessening symptoms while maintaining health systems. Building from an analysis of the therapeutics market landscape, the goal of this paper is to envision near- and medium-term actions the US government can take to accelerate access to the most promising COVID-19 treatments in lower-income countries as a crucial offensive against COVID-19. Overall, many of these actions will require additional investment on the part of the US government.
2. Understanding the Products: Issues and Unknowns
Several oral antiviral treatments have been shown to reduce morbidity and mortality, and improve recovery in clinical trials. This paper focuses on the two oral antiviral products with emergency use authorizations and the most advanced and actionable evidence.
2.1 Safety and Efficacy
Nirmatrelvir/r has been shown to reduce the risk of hospitalization and death by 89 percent when given to unvaccinated people within five days of symptom onset.[7] The drug seems to remain effective against Omicron based on in-vitro data.[8] The NIH recommends Paxlovid over remdesivir, molnupiravir, and monoclonal antibodies.[9] Pfizer is currently running a trial to assess the drug’s ability to block transmission in households, with results expected soon.
Molnupiravir was found to reduce the risk of hospitalization and death in high-risk patients by 30 percent.[10] The drug also appears to be effective against Omicron based on in-vitro data.[11] The US and UK granted emergency authorization in late 2021, while France has declined Merck’s emergency use application and India has excluded the drug from its national treatment protocol due to safety concerns.[12] NIH guidelines recommend only using molnupiravir to prevent high-risk patients from developing severe disease if Paxlovid, monoclonal antibodies, and remdesivir are unavailable.[13] Further studies will generate important information on efficacy in vaccinated individuals. As both nirmatrelvir/r and molnupiravir are new chemical entities, safety data is also still limited; active pharmacovigilance and monitoring are needed.
2.2 Treatment and Testing Guidelines
On March 3, 2022, WHO guidelines were updated to include a conditional recommendation for molnupiravir to be provided only to non-severe COVID-19 patients with the highest risk of hospitalization.[14] For nirmatrelvir/r, the WHO continues to evaluate evidence and has yet to make any recommendations. In the US context, the government announced a “test to treat” program that provides free access to Paxlovid for patients who test positive (though funding for this was stalled with the removal of the COVID supplemental from the recent US spending package).[15] Positive results of viral tests are required to prescribe both nirmatrelvir/r and molnupiravir in the US. Both also have drug-drug interactions (DDIs), which need to be managed carefully. Given limited testing infrastructure and DDI management capacity in resource-constrained settings, testing and DDI management requirements will likely be an impediment to oral antiviral access. In March 2022, the WHO released new guidance on rapid diagnostic tests for self-testing.[16] Further investment in diagnostic manufacturing and distribution will be crucial moving forward. In sub-Saharan Africa, only 1 in 20 people have been tested for COVID-19, and there are often delays between testing and result availability.[17] To the extent possible, investments in new diagnostic capabilities should be integrated in health systems strengthening both to support urgent response efforts and to guard against future health threats. Further work is needed on how to best incorporate antiviral treatments for COVID into primary healthcare delivery.
Lowest-tier pricing for bilateral deals with LMICs has not been made publicly available (discussed in more detail in section 4). The prices associated with these treatments will have significant implications for the value-for-money and cost-effectiveness calculations of oral antivirals and appropriate treatment protocols in different settings—each with varying levels of vaccination and immunity, testing capacity, and resource availability.
Table 1. Comparative efficacy, safety, and modality of key oral antivirals for COVID-19
|
|
Nirmatrelvir/ritonavir |
Molnupiravir |
|
Efficacy |
High |
Low |
|
Relative risk reduction among unvaccinated |
89% |
30% |
|
Absolute risk among unvaccinated |
7% to 1% |
9.7% to 6.5% |
|
Number needed to treat (NNT) |
17 |
31 |
|
Feasibility of administration |
Oral regimen, twice per day for five days |
Oral regimen, four capsules every 12 hours for five days |
|
Disease severity |
FDA authorized for treatment of mild-to-moderate COVID among those at high risk of progression to severe COVID and aged 12 and older |
FDA authorized for treatment of mild-to-moderate COVID among those at high risk of progression to severe COVID and aged 18 and older |
|
Initiation |
Within five days of symptom onset |
Within five days of symptom onset |
|
Safety |
Potentially significant drug-drug interactions
Risk of HIV-1 drug resistance
Ritonavir studied (safe) in pregnancy |
Not anticipated to have drug interactions
Not recommended for use during pregnancy, among other safety concerns |
Sources: FDA 2021a and 2021b; Harvard Medical School.
3. Understanding the Manufacturing Market: Issues and Unknowns
Given the range of bottlenecks, barriers to entry, demand inefficiencies, and gaps in testing infrastructure, there is considerable risk that production and uptake of oral antivirals will be far too slow and modest in scale. Based on interviews and analysis, we estimate that manufacturers for nirmatrelvir/r will begin filing for marketing authorization in LMICs—following approval—in 9 to 12 months under the best-case scenario, but will more realistically take up to 18 months (see timeline below) unless sufficient policy actions are taken. Therefore, the short-to-medium term supply outlook depends crucially on availability from Pfizer.
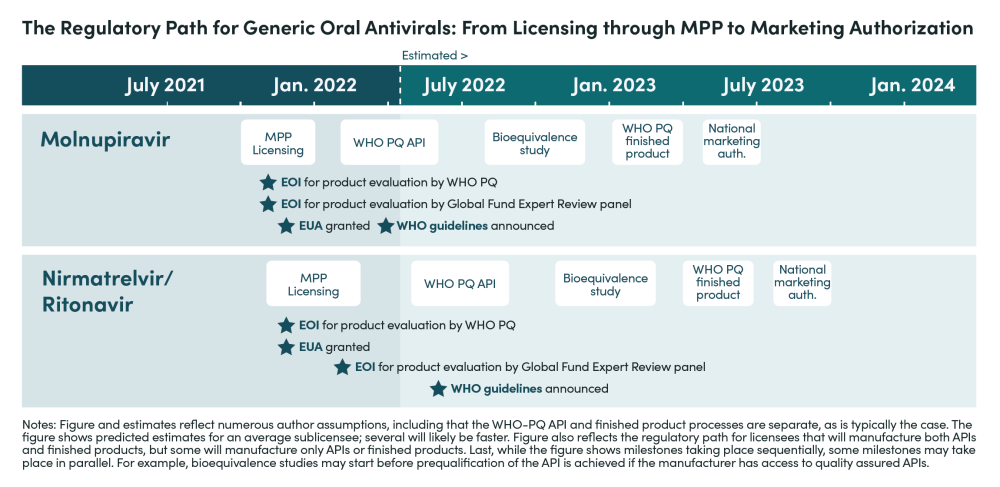
The key steps for manufacturers to complete before filing for market authorization in LMICs include:
-
Securing and completing API and finished product development and process validation: Without technical support from Pfizer, this could take between three and six months. This step could take longer if manufacturers have to wait for WHO prequalified APIs to become available.
-
Completing bioequivalence studies and putting together the relevant dossiers: Bioequivalence studies must be completed in vivo, not in vitro, as per the guidance released by WHO, and will involve receiving approval from regulatory agencies and ethics review boards and then conducting and finishing studies. This could also take three to six months. As of April 2022, Pfizer’s Paxlovid is under assessment for WHO PQ and two generic manufacturers have at least a preliminary dossier submitted.
-
Receiving WHO PQ: Prequalification is normally given up to 9 months after dossier submission for a product. This includes the time manufacturers take to provide any additional information requested by the WHO PQ team or address other concerns identified during the process. While submissions to WHO-PQ-EUL for molnupiravir and nirmatrelvir/r to date signal that at least some of the manufacturers who receive voluntary licenses will submit their dossiers quickly, assessment delays suggest that dossier submission alone may not be a reliable time epoch for anticipating product WHO-EUL and launch.
Companies without WHO PQ experience might require longer timeframes and additional support for successful PQ. If Global Fund Emergency Review Panel-based purchasing takes place, waiting on WHO-PQ EUL may not be needed for Global Fund procurement.
This section looks into the issues associated with each of these steps.
3.1 Promises and Pitfalls of Voluntary Licensing Arrangements
Voluntary licensing arrangements are a welcome modality to increase access to innovator products in LMICs, but translating licenses into access to medicines will require several more steps). Merck and Pfizer signed voluntary licensing agreements with the Medicines Patent Pool (MPP) for the manufacturing and supply of molnupiravir and nirmatrelvir/r in 105 and 95 low- and-middle-income countries (LMICs) in October and November 2021 and announced the sublicensees in January and March 2022, respectively. In addition, Merck bilaterally granted voluntary licenses to five Indian generic companies in March/April 2021. These licenses were signed before results from phase 3 clinical trials were available and the products were registered.
This effort to increase oral antiviral access to LMICs is good news, but has three main limitations: (1) the limited supply of APIs and Key Starting Materials/Intermediates could prevent licenses from translating into increased supply in the short term; (2) too many sub-licensees might lead to reduced interest and investments from generic manufacturers in configuring capacity and carrying out bioequivalence and other studies where required; and (3) licenses do not cover all populations in LMICs. Below, we discuss each in more detail.
1. API supplier availability: Once generic manufacturers are selected by MPP to sign sublicenses, they need to secure sufficient sources of APIs. These sources could either be developed and selected by the licensee manufacturers themselves (e.g., 18 companies were selected by MPP to produce the API for molnupiravir) or, in some cases, the licensee manufacturer could source from the API suppliers developed by the originators (i.e., Merck and Pfizer). In any case, and according to the license agreement, API suppliers must be prequalified by the WHO—which requires a physical inspection by the WHO or a member of the Pharmaceutical Inspection Co-operation Scheme[18] at some point in the last three years. Most generic API manufacturers are located in India and China, with a handful of suppliers located in Indonesia, South Africa, South Korea, and Pakistan. Strategic support to expand and streamline the pool of API and intermediate manufacturers—including robust signals of future demand, risk-sharing mechanisms, or subsidies in the form of push funding and technical support to increase yields—could help reduce the time for licensed manufacturers to submit their dossier to WHO-PQ and, in turn, the time to register and sell their products in LMIC markets.
2.Number of licenses: MPP is providing voluntary licenses for the manufacturing of oral antivirals to a large number of manufacturers. Between 2010, when MPP was created, and 2020, MPP secured licenses for 17 products for HIV, Hep C, and TB, and only signed sublicenses with 23 generic manufacturers and product developers.[19] In contrast, MPP is signing over 20 licenses for each oral antiviral. On January 20, 2022, MPP announced that 27 generic manufacturers from 11 Asian and African countries[20] had been selected to sign sublicense agreements to produce the raw ingredients for molnupiravir (5 companies), the finished product itself (9 companies), or both (13 companies). Adding to Merck’s bilateral voluntary licensing agreements with 5 Indian manufacturers from April 2021, this announcement brought the number of generic manufacturers with licenses for molnupiravir to 32. On March 17, 2022, MPP announced that 35 generic manufacturers from 12 countries[21] had been selected to produce the raw ingredient for nirmatrelvir (6 companies), the finished product (9 companies), or both (20 companies).
The companies selected by MPP for voluntary licenses are geographically diverse, which could enable greater supply chain adaptability and flexibility in the face of potential disruptions. While signing many licenses may reduce geographical concentration and increase supply and competition, it also increases the burden on already-stretched regulatory authorities. These agencies may struggle to provide adequate pre- and post-marketing quality assurance (see next point on regulatory challenges). Further, the presence of dozens of licensed manufacturers in the market might deter companies from investing in manufacturing capacity amid uncertain demand and profitability prospects. For example, Gilead’s remdesivir voluntary license program included nine manufacturers and enabled access to only 2.3 million people in LMICs in the first year of operation, though it now reports reaching 7 million people in LMICs.[22] It is still too soon to determine the real impact of the large number of licenses and generic manufacturers on competition and supply, but as of February 14, 2022, only 12 companies had initiated the WHO prequalification process for molnupiravir (6 companies had early interactions with WHO on prequalification of finished products, 3 submitted dossiers for API prequalification that WHO accepted for assessment, and 3 did both).[23] An even smaller number will market their products—the final step to translate licensing into increased access.
3. Country reach: Voluntary licensing agreements exclude many upper-middle-income countries, including countries with established manufacturing capacity such as Argentina, Bulgaria, and Colombia. Those countries will have to rely on supply from Pfizer and Merck, but as more high-income countries sign advance purchase agreements, they are likely to be at the back of the delivery queue.
3.2 Regulation: A Key Step to Fast-Track Access to Quality-Assured Oral Antivirals in LMICs
Securing an adequate supply of new oral antivirals will require meeting relevant country and global regulatory standards. In theory, manufacturers could follow different regulatory pathways (see Figure 1). Still, in reality, licensed manufacturers will have to obtain regulatory approval from WHO PQ or a stringent regulatory authority (SRA) before applying for marketing authorization in LMICs where the products will be sold. This clause, explicit in the voluntary licensing agreements between Merck and Pfizer and MPP, is aligned with existing requirements from international procurement organizations. The WHO estimates that only 30 percent of National Regulatory Authorities (NRAs) among its member states have the capacity to regulate medical products effectively and efficiently in their countries.[24]However, there is not currently a pathway for SRAs to assess generic COVID-19 products intended to be marketed only outside their jurisdictions, and regulatory decisions by functional NRAs in LMICs such as Tanzania, Nigeria, Egypt, and Ghana are not recognized. The resulting centralization of all regulatory assessments within WHO PQ risks extending regulatory timelines and delaying access, among other negative consequences. Proceeding without a post-marketing surveillance system in place (the WHO PQ focuses on market entry) and equating WHO PQ status with marketing authorizations in LMICs also hinder timeliness and access.
Figure 1. Registration pathways for generic oral antivirals
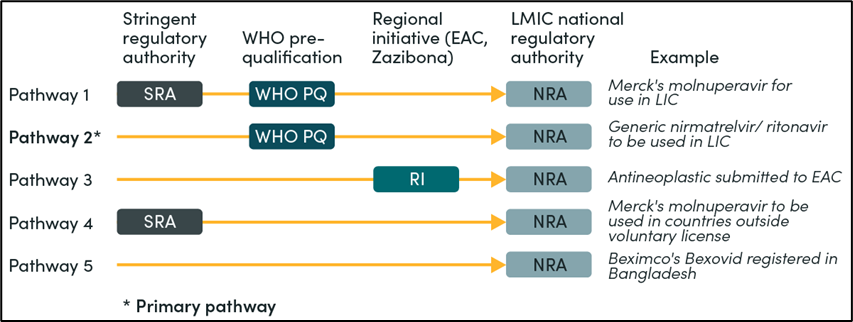
Accelerating the regulatory process for oral antivirals in LMICs will require: (1) expanded regulatory capacity beyond the WHO PQ; (2) better coordination and alignment between the WHO PQ program and NRAs; and (3) additional support to manufacturers of generic products.
1. Expanded regulatory capacity: Stringent regulatory authorities could expand existing programs to review marketing applications for medicines intended to be marketed outside their jurisdictions to increase the capacity of the WHO PQ program. For example, the US FDA could incorporate oral antivirals for the treatment of COVID-19 into its existing PEPFAR process to review marketing applications of HIV medicines (included in our recommendations below). Products assessed by the US FDA are added to the list of medicinal products prequalified by WHO, expanding the regulatory capacity of the WHO PQ program for HIV. Currently, 86 medicines or around 30 percent of the antiretrovirals (ARVs) in the WHO PQ list were included based on assessments and inspections conducted by the US FDA. A similar outcome is desirable for oral antivirals to treat COVID-19.[25]
2. Coordination and alignment: Prequalified products still need to be approved by the NRAs of the countries in which market entry is sought. In response to this challenge, the WHO created the WHO Collaborative Procedure to reduce duplication and shorten time to registration. As part of the procedure, NRAs receive access to PQ assessments and inspection outcomes, and in return, they commit to reaching a marketing authorization decision within 90 days. Although the scheme has grown and now has around 50 participating NRAs, national legal frameworks and regulatory procedures are still a barrier to the scheme; many NRAs lack adequate legal provisions to rely on external data and assessments.
3. Support to manufacturers of generic products: Pfizer can facilitate requisite manufacturing and regulatory processes for sublicensees. For instance, Pfizer could establish a comprehensive technology transfer program to share all information needed to develop and manufacture companies’ own versions of the product. Merck and Pfizer could also allow licensed manufacturers to use their own authorized suppliers of API and excipients.
To overcome short-term supply constraints and accelerate availability, Merck and Pfizer could also designate some generic producers to act as their contract manufacturers (i.e., secondary manufacturing sites) and agree on how manufacturing outputs would be split between high-income country markets and LMIC markets. This would reduce the time required for licensed manufacturers to achieve commercial-grade production (i.e., licensed manufacturers would produce using the same manufacturing process, ingredients, quality controls, etc.) while also removing the need to conduct in-vivo bioequivalence studies. For this strategy to work, Merck and Pfizer would need to include these manufacturing sites within their own submission to WHO PQ and LMICs NRAs. While this might be possible, Merck and Pfizer are expecting licensed manufacturers to have full responsibility and authority over the development, registration, importation, manufacture, and commercialization under the licensing agreement.
3.3 Implications for the Production of Ritonavir to Treat HIV/AIDS
Ritonavir (RTV), one of the ingredients used in the new oral antiviral developed by Pfizer, is manufactured by nine generic manufacturers under voluntary licensing agreements signed between Abbvie and MPP in 2014 and 2015.[26]The medicine, introduced as a first-line treatment of HIV in the 1990s, is now only recommended by the WHO in combination with other ARVs as a second-line treatment for adults and adolescents or as an alternative first-line option for children. Around 2 million patients annually are estimated to be on RTV-based combinations globally based on these WHO recommendations.
Additionally, RTV-based combinations are currently used in around 32 countries that have not yet implemented the WHO recommendation to transition to dolutegravir-based regimens as first-line therapy for HIV/AIDS.[27] The transition started in 2016—after clinical trials showed that dolutegravir-based regimens were better tolerated and more convenient, effective, and protective against resistance than previous regimes using RTV and other ARVs—and has accelerated worldwide in the past 5 years. Estimates suggest 6 to 9 million people had access to generic dolutegravir in 2019, and 82 LMICs were reported to be transitioning to dolutegravir-based regimens. By mid-2020, according to the WHO, transition to dolutegravir had been implemented in 100 LMICs. At present, there are approximately one million adults on RTV-containing ARVs in LMICs, and the number continues to decline as additional patients transition to dolutegravir (DTG). Still, some patients will need to stay on RTV-containing treatments, in case of failure or intolerance to DTG. An estimated 5 percent of children living with HIV will need to continue receiving lopinavir/r treatment due to DTG intolerance. In total, roughly 30 million RTV-containing tablets were needed in 2020.[28]
As more countries follow WHO guidance and implement DTG-based regimens, reduced demand for fixed-dose combinations with RTV will ease supply pressure on the availability of RTV API and finished tablets for nirmatrelvir/r, but this shift will require careful coordination. The nine manufacturers of RTV-based combinations for the treatment of HIV have already secured WHO PQ or FDA tentative approval and therefore comply with regulatory standards for one of the nirmatrelvir/r active ingredients. These companies effectively have a head start with production of nirmatrelvir/r. Generic manufactures thus may deprioritize RTV for HIV patients in their production and campaign planning due to small order sizes and configure their production towards nirmatrelvir/r, which could contribute to longer lead times for RTV-containing products for HIV patients. PEPFAR, the Global Fund, South Africa, and other ARV purchasers might need to place early orders for RTV for HIV and even stockpile some treatments to avoid dangerous delays.
3.4 Indemnity and Liability Issues
Indemnity and liability challenges should be tackled early and comprehensively. Oral antivirals, delivered only to people diagnosed with COVID rather than healthy individuals, will be administered at significantly lower rates than COVID vaccines. Nonetheless, molnupiravir and nirmatrelvir/r are new chemical entities for which the research and development process is incomplete. Both have received emergency use authorizations, as opposed to full approval, from regulators; some uncertainty remains about the potential risks of these products. Manufacturers and stakeholders interested in selling, buying, or donating them in LMICs are likely to be concerned about legal exposure should adverse events arise. In response to this challenge, the licensing agreements between Merck and Pfizer and MPP require licensed manufacturers to maintain comprehensive liability insurance and specify that licensees are solely responsible for any liability associated with APIs and finished products.
4. Understanding the Procurement Landscape: Issues and Unknowns
4.1 Demand-Side Considerations
Demand-side challenges for therapeutics are more significant than vaccines. The pathway from regulatory approval/EUL to clinician acceptance and patient uptake is much more complex for therapeutics. As discussed, licensing alone does not provide sufficient incentives for manufacturers to build new or reconfigure existing capacity. Uncertain future demand for other COVID-19 technologies has posed obstacles to production scale-up. Policymakers can employ a range of tools to organize and signal demand to manufacturers. These include volume guarantees, technology transfers, production or capacity subsidies, concessional loans, and grants.[29] The right combination of financial and technical instruments to encourage sufficient capacity expansion requires further analysis, especially to understand the effects of existing procurers (or manufacturers) competing for supply. The ability of an Advance Market Commitment (AMC) to drive down prices in the case of Paxlovid is unclear. However, an AMC-like guarantee (i.e., a signal of earmarked funding to purchase Paxlovid) may help achieve greater capacity investments by some high-quality generic producers. To this end, $200-300 million appears to be a reasonable amount to earmark and signal.
These resources may come from a range of buyers. Procurement in LMIC markets involves three main segments, each with its own set of vagaries:
-
Private market in some LMICs, where patients pay out of pocket: In this market segment, WHO-PQ is not required, and multiple generic manufacturers can sell. Out-of-pocket buyers face uncertain medicines quality and variable prices.[30] In such settings, establishing a test-treat approach is difficult.
-
Public procurement-led market in which LMIC governments purchase health products with domestic resources (including World Bank financing channeled through public coffers): This market segment also does not technically require manufacturers to have WHO-PQ. However, there is more visibility into pricing and greater scope to set up test-treat programs, especially amid recently released WHO guidance on rapid diagnostic tests for self-testing.[31]
-
Global procurement market where multilateral organizations pool resources and purchase products for LMICs: Manufacturers are required to have WHO-PQ to sell through global procurement mechanisms. Global procurement agents can presumably obtain the lowest tier pricing from the originator. They can also help create robust test-treat programs in-country. The remaining central uncertainty is what financing and procurement modality will be used. In the case of the Global Fund, existing resources in the COVID-19 Response Mechanism could be quickly reprogrammed to purchase oral therapeutics, followed by new resources for additional procurement.
Given that pooled donor financing will likely make up the greatest share of the oral antivirals market in LMICs, demand-side uptake of oral antivirals in LMICs depends critically on WHO recommendations and guidelines for their use. These resources are updated regularly as new evidence emerges. Many LMICs will wait to include new oral therapeutics in their national treatment guidelines until the WHO guidelines recommend or conditionally recommend the use of a therapeutic.
While many LMIC officials are closely following the US test-to-treat program, they are awaiting WHO recommendations on nirmatrelvir/r before adding treatment protocols to their national guidelines. Similarly, agencies that might provide global financing for oral antivirals (e.g., the Global Fund and the ACT-A therapeutics pillar) have also held off announcing or releasing grant funding until WHO recommendations are in place. This creates significant demand-side uncertainty for financiers, procurers, and manufacturers.
4.2 Relevant Deals and Pricing
The US has agreed to a deal for 20 million courses of Paxlovid priced at $530 per treatment course, seemingly intended for domestic use.[32] The US also agreed to a $2.2 billion deal for 3.1 million courses of molnupiravir priced at $700 per treatment course, now all delivered.[33] Pfizer plans to produce at least 120 million courses of Paxlovid this year, of which 30 million courses have already been sold to mostly high-income countries. Pfizer is also expected to provide 10 million courses to LMICs via the Global Fund at an unspecified price.[34] Pfizer also plans to supply up to 4 million courses of nirmatrelvir/r through UNICEF to low-income countries at a non-profit price and to upper middle-income countries under a tiered pricing system. With delivery slated to start in April 2022 and continue through the end of the year, the goal is to provide short-term access while the licensed manufacturers selected by MPP prepare for production. [35] UNICEF will work closely with the WHO and other ACT-A partners to ensure equitable access to the volume under this agreement.
Further, the Africa Centres for Disease Control and Prevention (Africa CDC) has agreed to a memorandum of understanding with Pfizer for Paxlovid, though deal specifics have not yet been announced.[36] Details on lowest-tier pricing and whether African markets will be prioritized in the delivery “queue” given lower vaccination rates remain to be seen. Clearer expectations of global access pricing and production cost estimates would be valuable, even as supply and demand estimates would be expected to shift over time.
5. Priority Actions for US Policymakers
[Note: Follow-on work from CGD in the coming year will offer recommendations to other entities in the global health architecture based on further analysis.]
-
The US government should develop a global oral antiviral access strategy. With adequate resources, the strategy would first and foremost organize and generate demand. Once the demand side appears more certain, it will enable greater upfront investments in manufacturing capacity. Given that short-term access in LMICs depends on Pfizer and Merck’s production, the strategy should incorporate incentives for them to further scale-up manufacturing to the extent possible. With confirmed advance purchase orders for LMICs, the Global Fund can also signal a stronger demand-side market. In addition to originator production incentives, the strategy should also incorporate a testing plan that harnesses the capacities and country reach of PEPFAR and PMI and an approach to stockpiling, which could involve price contingencies dependent on use.
-
USAID should leverage its implementing partner architecture to help create country deployment programs for test-treat initiatives, building on the suggested oral antiviral access strategy. The US domestic test and treat strategy involves identifying ways to manage the complexities of DDIs for some of the oral antivirals. These efforts should be adapted quickly for use in resource limited health systems. PEPFAR and PMI implementers who have significant experience in supporting HIV, malaria, and TB test and treat in LMICs, especially African countries, could then be resourced to initiate testing and treatment programs by providing countries the relevant tools and technical resources. Implementing test-treat requires coordination with test manufacturers, procurers, pharmaceutical companies (generic and originator), and systems to translate co-administration advice into field settings.[37]
-
USAID should leverage its implementing partner architecture and partnership with US Pharmacopeia (USP) to further enhance technical support programs for generic manufacturers who have received voluntary licenses directly or through MPP. Such assistance would help speed up the timelines for voluntary licensees to submit to WHO PQ. As investments in process intensification and new formulation pathways yield results, this technical assistance program could help disseminate and spur more rapid use of such developments to multiple VL manufacturers. Technological support models that facilitate the innovation process in process intensification for API and finished product manufacturing should build from and leverage ongoing efforts funded outside the USAID ecosystem.
-
The US government should initiate a close partnership with Pfizer and Merck to ensure needs-based allocation of limited supplies of oral antivirals by leveraging the deployment plans above. Such a partnership should define a single point of interface for the companies across different agencies working on COVID therapeutics.
-
The FDA should expand the conditional approval pathway currently used for HIV medicines to include generic oral antivirals for COVID-19 to be delivered outside of the US. The FDA normally takes around six months to review a product under the PEPFAR scheme. Leveraging similar approval pathways would increase global regulatory capacity to assess oral antivirals manufactured by companies with voluntary licensing agreements with MPP, Pfizer and Merck, strengthen the WHO PQ program, and reduce the time for products to reach LMIC markets. Additional support may be required for the FDA to dedicate resources towards this effort. As part of the existing PEPFAR process, numerous procedures and guidelines have already been developed and published. Licensed generic manufacturers are also familiar with the program and have used it extensively.[38]
More broadly, the FDA, the WHO, and manufacturers should take this opportunity to enhance their information sharing and collaboration. For example, the products approved by the FDA could be incorporated in the WHO Collaborative Procedure to speed up registration by NRAs in LMICs. PEPFAR and WHO PQ databases could also make the same information available; the PEPFAR database currently does not report information on APIs or include public assessments or inspection reports. -
The US government should support regional and national health technology assessments—through technical support from USAID and engagement with global health multilaterals—to determine the costs and benefits associated with introducing different oral antivirals and to inform spending decisions. Given LMIC variation in vaccination coverage, seroprevalence and natural immunity, testing capabilities, and fiscal space, as well as some remaining uncertainty on efficacy, safety and price, the US government should encourage global procurement mechanisms to deploy oral antivirals where economic evaluations of multiple oral antivirals have been conducted and the value for money is greatest. Demand and funding for oral antivirals are likely to compete with other health care technologies, including vaccines, diagnostics, and PPE. It is essential to maximize health gains in global and country-level policy decisions, especially given current fiscal constraints.
-
The US government should create a consultative procurement forum with Africa CDC and the ACT-A therapeutics and diagnostics pillars, including UNITAID, FIND, the WHO, the Global Fund, UNICEF Supply Division, and PAHO. This platform would facilitate information exchange and help ensure that oral antiviral market dynamics are sufficiently coordinated amid agreement on core principles. “Market health” must be given due importance in procurement awards by each of the procurement agencies. An existing external advisory group to the ACT-A therapeutics pillar management team could also be the right forum to discuss evolving demand forecasts.
-
The US government should urge the World Bank to help countries secure no-fault compensation programs for those who suffer serious adverse events associated with the administration of oral antiviral treatments through their programs. Setting up this program as early as possible will prevent the situation experienced in several COVAX-supported countries where legal liability issues associated with COVID vaccines slowed or halted product rollout.
[1] Our World in Data. “Coronavirus (COVID-19) Vaccinations.” https://ourworldindata.org/covid-vaccinations.
[2] Tada, Takuya, et al. “Comparison of Neutralizing Antibody Titers Elicited by mRNA and Adenoviral Vector Vaccine against SARS-CoV-2 Variants.” Frontiers in Immunology (March 8, 2020). https://www.frontiersin.org/articles/10.3389/fimmu.2022.797589/full.
[3] Gottlieb, Robert L., et al. “Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients.” The New England Journal of Medicine 386 (January 27, 2022): 305-315. https://www.nejm.org/doi/full/10.1056/NEJMoa2116846?query=featured_home.
[4]Medicines Patent Pool. "35 generic manufacturers sign agreements with MPP to produce low-cost, generic versions of Pfizer’s oral COVID-19 treatment nirmatrelvir in combination with ritonavir for supply in 95 low- and middle-income countries.” March 17, 2022. https://medicinespatentpool.org/news-publications-post/35-generic-manufacturers-sign-agreements-with-mpp-to-produce-low-cost-generic-versions-of-pfizers-oral-covid-19-treatment-nirmatrelvir-in-combination-with-ritonavir-for-supply-in-95-low-and.
[5] Medicines Patent Pool. “27 generic manufacturers sign agreements with MPP to produce low-cost versions of COVID-19 antiviral medication molnupiravir for supply in 105 low- and middle-income countries.” January 20, 2022. https://medicinespatentpool.org/news-publications-post/27-generic-manufacturers-sign-agreements-with-mpp-to-produce-molnupiravir.
[6] Merck. “Amid Humanitarian Crisis in India, Merck Announces Voluntary Licensing Agreements with Five Indian Generics Manufacturers to Accelerate and Expand Global Access to Molnupiravir, an Investigational Oral Therapeutic for the Treatment of COVID-19.” April 27, 2021. https://www.merck.com/news/amid-humanitarian-crisis-in-india-merck-announces-voluntary-licensing-agreements-with-five-indian-generics-manufacturers-to-accelerate-and-expand-global-access-to-molnupiravir-an-investigational-ora/.
[7] Hammond, Jennifer, et al. “Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19.” The New England Journal of Medicine (February 16, 2022). https://www.nejm.org/doi/full/10.1056/NEJMoa2118542.
[8]Pfizer. “Pfizer Shares In Vitro Efficacy of Novel COVID-19 Oral Treatment Against Omicron Variant.” January 18, 2022. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-vitro-efficacy-novel-covid-19-oral-treatment.
[9] National Institutes of Health. “Therapeutic Management of Nonhospitalized Adults with COVID-19.” February 1, 2022. https://www.covid19treatmentguidelines.nih.gov/therapies/statement-on-therapies-for-high-risk-nonhospitalized-patients/.
[10] Merck. “Merck and Ridgeback Biotherapeutics Provide Update on Results from MOVe-OUT Study of Molnupiravir, an Investigational Oral Antiviral Medicine, in At Risk Adults With Mild-to-Moderate COVID-19.” November 26, 2021. https://www.merck.com/news/merck-and-ridgeback-biotherapeutics-provide-update-on-results-from-move-out-study-of-molnupiravir-an-investigational-oral-antiviral-medicine-in-at-risk-adults-with-mild-to-moderate-covid-19/.
[11] Merck. “Merck and Ridgeback’s Molnupiravir, an Investigational Oral Antiviral COVID-19 Medicine, Demonstrated Activity Against Omicron Variant in In Vitro Studies.” January 28, 2022. https://www.merck.com/news/merck-and-ridgebacks-molnupiravir-an-investigational-oral-antiviral-covid-19-medicine-demonstrated-activity-against-omicron-variant-in-in-vitro-studies/.
[12] Spencer, Mimosa and Emilio Pardoi. “France cancels order for Merck’s COVID-19 antiviral drug.” Reuters, December 22, 2021. https://www.reuters.com/world/europe/france-cancels-order-mercks-covid-19-antiviral-drug-2021-12-22/; Das, Krishna N and Ankur Banerjee. “India says safety concerns restricting use of Merck COVID pill.” Reuters, January 5, 2022. https://www.reuters.com/world/india/india-health-official-says-merck-covid-pill-has-major-safety-concerns-2022-01-05/?rpc=401&.
[13] National Institutes of Health. “Therapeutic Management of Nonhospitalized Adults with COVID-19. February 1, 2022. https://www.covid19treatmentguidelines.nih.gov/therapies/statement-on-therapies-for-high-risk-nonhospitalized-patients/.
[14] World Health Organization. “WHO updates its treatment guidelines to include molnupiravir.” March 3, 2022. https://www.who.int/news/item/03-03-2022-molnupiravir; Agarwal, Arnav, et al. “A living WHO guideline on drugs for covid-19.” British Medical Journal 370 (2020). https://www.bmj.com/content/370/bmj.m3379.
[15] As of March 2022, the drugs are not authorized in the US for: use longer than five consecutive days; initiation of treatment in patients requiring hospitalization due to severe or critical symptoms; or pre- or post-exposure prophylaxis.
[16] World Health Organization. “Use of SARS-CoV-2 antigen-detection rapid diagnostic tests for COVID-19 self-testing.” March 9, 2022. https://www.who.int/publications/i/item/WHO-2019-nCoV-Ag-RDTs-Self_testing-2022.1.
[17] Wroe, Emily B., et al. “Test and treat: a missing link in the global fight against COVID-19.” The Lancet Global Health 10, no. 2 (February 1, 2022): E181-182. https://www.thelancet.com/journals/langlo/article/PIIS2214-109X(21)00568-4/fulltext.
[18] A list of authorities participating in the Pharmaceutical Inspection Co-operation Scheme is available here: https://picscheme.org/en/members
[19] Medicines Patent Pool. “MPP’s Brochure – Expanding access to public health.” September 7, 2021. https://medicinespatentpool.org/news-publications-post/mpp-s-brochure.
[29] Bangladesh, China, Egypt, Jordan, India, Indonesia, Kenya, Pakistan, South Africa, South Korea, and Vietnam.
[21] Bangladesh, Brazil, China, Dominican Republic, Jordan, India, Israel, Mexico, Pakistan, Serbia, South Korea, and Vietnam.
[22] Gilead. “Gilead Sciences Announces Step to Expand Availability of Remdesivir in India.” April 26, 2021. https://www.gilead.com/news-and-press/press-room/press-releases/2021/4/gilead-sciences-announces-steps-to-expand-availability-of-remdesivir-in-india; Gilead. “FDA Approves Veklury (Remdesivir) for the Treatment of Non-Hospitalized Patients at High Risk for COVID-19 Disease Progression.” January 21, 2022. https://www.gilead.com/news-and-press/press-room/press-releases/2022/1/fda-approves-veklury-remdesivir-for-the-treatment-of-nonhospitalized-patients-at-high-risk-for-covid19-disease-progression.
[23] World Health Organization. “Status of COVID-19 Medicines and Active Pharmaceutical Ingredients (APIs).” February 14, 2022. https://extranet.who.int/pqweb/sites/default/files/documents/COVID-19_PQ_Tracking_14Feb2022.pdf.
[24] World Health Organization. “WHO Essential Medicines & Health Products Annual Report 2017: Towards Access 2030.” 2018. https://apps.who.int/iris/bitstream/handle/10665/272972/WHO-EMP-2018.01-eng.pdf.
[25] In the long-term, Pfizer and Merck could consider upgrades to existing formulations to increase adherence and absorption to improve patient acceptability and reduce regulatory requirements for in-vivo bioequivalence studies. For example, if Merck decided to reformulate molnupiravir from a 200 mg capsule to an 800 mg tablet, a patient would only need to take 10 tablets instead of 40 to treat COVID. At the same time, an improvement in absorption, solubility, and permeability would change the bioequivalence requirement from an in-vivo bioequivalence study to an in-vitro biowaiver.
[26] Adcock Ingram, Arene Lifesciences Ltd, Aurobindo Pharma Ltd, Emcure Pharmaceuticals Limited, Desano, Hetero Labs Limited, Lupin and Sun Pharmaceutical Industries Limited
[27] The Lancet HIV. “End resistance to dolutegravir roll-out.” September 2020. https://www.thelancet.com/action/showPdf?pii=S2352-3018%2820%2930231-9.
[28] Stakeholder interview with Clinton Health Access Initiative
[29] Kazaz, Burak, Scott Webster, and Prashant Yadav. “How Can We Encourage COVID-19 Vaccine Developers to Expand Manufacturing Capacity?” Center for Global Development. March 26, 2021. https://www.cgdev.org/blog/how-can-we-encourage-covid-19-vaccine-developers-expand-manufacturing-capacity.
[30]Silverman, Rachel, et al. “Tackling the Triple Transition in Global Health Procurement.” Center for Global Development. June 17, 2019. https://www.cgdev.org/sites/default/files/better-health-procurement-tackling-triple-transition.pdf.
[31] World Health Organization. “Use of SARS-CoV-2 antigen-detection rapid diagnostic tests for COVID-19 self-testing.” March 9, 2022. https://www.who.int/publications/i/item/WHO-2019-nCoV-Ag-RDTs-Self_testing-2022.1.
[32] Mishra, Manas and Erman Michael. “Pfizer’s COVID product sales to top $50 bln this year, investors want more.” Reuters, February 8, 2022. https://www.reuters.com/business/healthcare-pharmaceuticals/pfizer-forecasts-54-bln-2022-sales-covid-vaccine-pill-2022-02-08/.
[33] Merck. “Merck and Ridgeback Announce U.S. Government to Purchase 1.4 Million Additional Courses of Molnupiravir, an Investigational Oral Antiviral Medicine, for the Treatment of Mild-to-Moderate COVID-19 in At Risk Adults.” November 9, 2021. https://www.merck.com/news/merck-and-ridgeback-announce-u-s-government-to-purchase-1-4-million-additional-courses-of-molnupiravir-an-investigational-oral-antiviral-medicine-for-the-treatment-of-mild-to-moderate-covid-19-in-a/.
[34] Erman, Michael. “Pfizer to provide 10 mln courses of COVID pill to developing countries – the Global Fund.” Reuters, March 2, 2022. https://www.reuters.com/world/pfizer-provide-10-mln-courses-covid-pill-developing-countries-the-global-fund-2022-03-02/.
[35] Kimball, Spencer. “Pfizer to supply 4 million Covid antiviral treatments to poorer nations through UNICEF.” CNBC, March 22, 2022. https://www.cnbc.com/2022/03/22/pfizer-to-supply-4-million-covid-antiviral-treatments-to-poorer-nations-through-unicef.html
[36] Reuters. “Africa CDC has MOU with Pfizer for supplies of COVID-19 pill.” Reuters, March 10, 2022. https://www.the-star.co.ke/news/2022-03-10-africa-cdc-has-mou-with-pfizer-for-supplies-of-covid-19-pill/.
[37] As highlighted by the need for diagnostic capabilities, health systems strengthening should underpin and be brought to bear in efforts to respond to the current pandemic and prepare for future health shocks.
[38] This measure, through which the FDA can issue tentative approval for a product that is not yet be fully approved in the US due to patents or market exclusivities but meets all safety, efficacy, and quality requirements, has been in place since 2004 for generic ARVs to treat HIV/AIDS. It originally included PEPFAR and then expanded to the WHO PQ program since products assessed by the US FDA are added to the list of products prequalified by WHO. Currently, 86 medicines or around 30 percent of the ARVs listed in the WHO PQ list were included based on assessments and inspections conducted by the US FDA.
Topics
CITATION
Guzman, Javier, Julia Kaufman, and Prashant Yadav. 2022. Policy Actions for the US Government to Accelerate Access to Oral Antivirals for COVID-19 in Low- and Middle-Income Countries. Center for Global Development.DISCLAIMER & PERMISSIONS
CGD's publications reflect the views of the authors, drawing on prior research and experience in their areas of expertise. CGD is a nonpartisan, independent organization and does not take institutional positions. You may use and disseminate CGD's publications under these conditions.